03 May 2024 | By : Dr Mustafa Hashim
Carbon dioxide (CO₂), often perceived as a harmless gas, transforms into a potent antagonist in the presence of water, driving a detrimental process known as CO₂ corrosion or sweet corrosion (Fig.1). This insidious phenomenon wreaks havoc on metallic infrastructure across diverse industries, impacting pipelines, storage tanks, and boiler condensate systems. A thorough understanding of the intricate interplay between CO₂, water, and metal surfaces is paramount to mitigate this widespread damage mechanism.
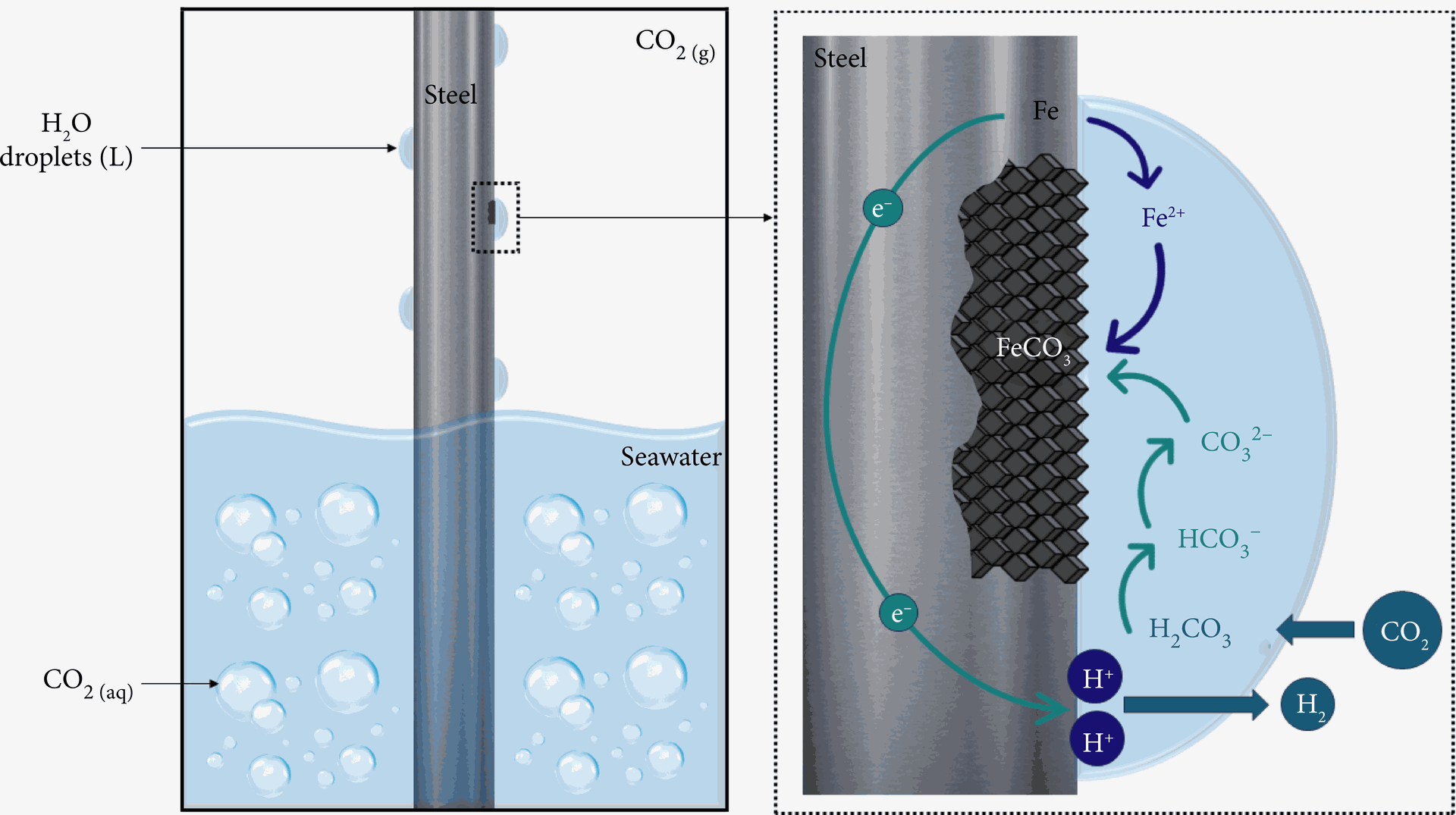
Fig.1: Schematic of CO2 corrosion in carbon steel. (source: Fonseca et al. (2024), International Journal of Corrosion https://doi.org/10.1155/2024/5537767 )
CO₂ Dissolution: From Gas to Acid
The transformation of CO₂ into carbonic acid (H₂CO₃) within an aqueous environment occurs through a series of well-defined chemical reactions. While CO₂ exhibits acidic properties when dissolved in water, the dominant species at different pH levels is not simply H⁺. The key concept here is acid-base equilibria. CO₂ acts as a weak diprotic acid, meaning it can donate two protons (H⁺) in a stepwise manner. These equilibria are represented by the following dissociation reactions:
- Hydration:
CO₂ (g) + H₂O (l) ⇌ H₂CO₃ (aq) [Eq.1]
(K₁ ≈ 4.45 x 10⁻⁷ M atm⁻¹ at 25 °C).
- Dissociation:
H₂CO₃ (aq) ⇌ H⁺ (aq) + HCO₃⁻ (aq) [Eq.2]
(K₂ ≈ 4.69 x 10⁻³ at 25 °C)
HCO₃⁻ (aq) ⇌ H⁺ (aq) + CO₃²⁻ (aq) [Eq.3]
(K₃ ≈ 4.76 x 10⁻¹¹ at 25 °C)
Here, K₁ and K₂ are the respective equilibrium constants for each dissolution and dissociation steps. These constants indicate the propensity of the reaction to proceed towards products (dissociation) or reactants (reassociation).
These dissociation steps establish the acidic nature of the solution. The extent of dissociation is highly dependent on the prevailing pH (Fig.2), as will be discussed below.
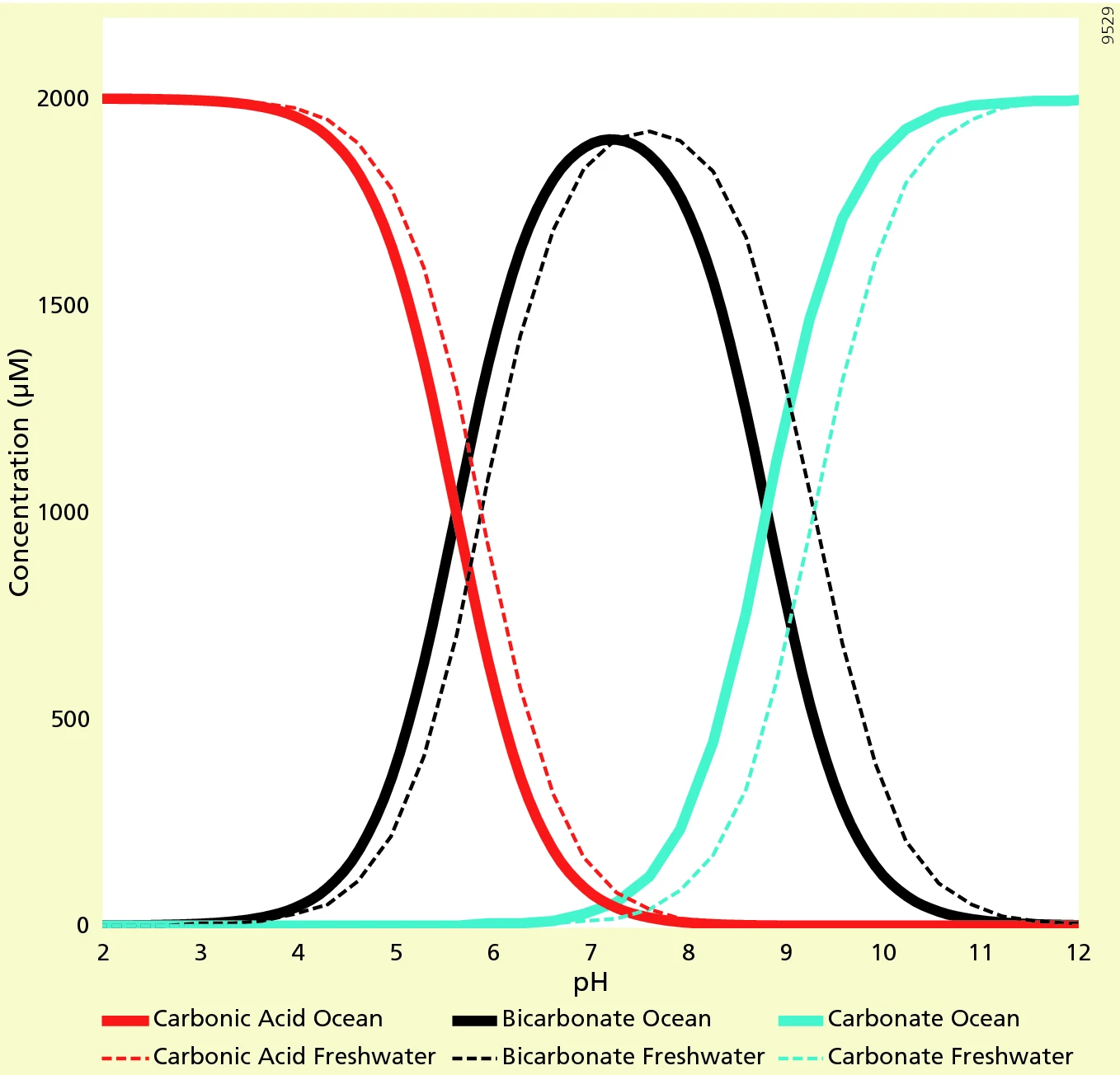
Fig.2: The distribution of carbonate species in fresh water and seawater as function of pH. (Source J.J. Middelburg, Biogeochemical processes and inorganic carbon dynamics, 2019,
https://doi.org/10.1007/978-3-030-10822-9_5).
Low pH (Acidic, pH < 7):
At low pH, there is a higher concentration of H⁺ ions in the solution. This high concentration of H⁺ drives the first dissolution step (hydration) of CO₂ towards the formation of H₂CO₃. However, the conversion of CO₂ to H₂CO₃ is limited due to the relatively small value of the equilibrium constant K₁.
Despite the limited conversion, some H₂CO₃ molecules still dissociate (second dissociation) to form H⁺ and HCO₃⁻ due to the presence of existing H⁺ ions. However, it's crucial to understand that this dissociation is limited as well. The equilibrium constant K₂ for the second dissociation signifies a dynamic equilibrium where both forward (dissociation) and backward (reassociation) reactions occur. Therefore, even though H⁺ originates from the dissociation of H₂CO₃, the limited extent of this dissociation ensures that the concentration of H₂CO₃ remains higher than the concentration of H⁺ at this acidic pH.
Neutral pH (pH ≈ 7):
At neutral pH, the concentration of H⁺ and HCO₃⁻ ions are approximately equal. In this balanced environment, the second dissociation step of carbonic acid (Eq.2) becomes particularly important. With the relatively low concentration of H⁺ compared to acidic conditions, the equilibrium between H₂CO₃ and HCO₃⁻ is balanced. This means that both the forward (dissociation of H₂CO₃) and backward (reassociation of H⁺ and HCO₃⁻) reactions occur at similar rates.
As a result of this balanced equilibrium and the limited conversion of CO₂ to H₂CO₃ due to the small value of K₁ (the equilibrium constant for the first dissociation), neither H₂CO₃ nor HCO₃⁻ completely dominates the system. However, the concentration of HCO₃⁻ becomes slightly higher than H₂CO₃ at neutral pH. This makes bicarbonate (HCO₃⁻) the dominant species in the CO₂-water system under these neutral conditions.
High pH (Alkaline, pH > 7):
At high pH (alkaline, pH > 7), the system undergoes a fascinating shift driven by the prevailing low concentration of H⁺ ions. This low H⁺ concentration, a hallmark of high pH itself, influences the dissociation of carbonic acid (H₂CO₃) in two crucial ways:
- Favourable Equilibrium Shift: The second dissociation step (Eq.2) becomes highly sensitive to the low H⁺ concentration. Le Chatelier's principle dictates that a system at equilibrium responds to changes by shifting in a way that counteracts the change. In this case, the low H⁺ concentration "pushes" the equilibrium towards the formation of more HCO₃⁻ ions. This essentially means more H₂CO₃ molecules dissociate to consume the limited H⁺ ions and establish a new equilibrium with a higher concentration of HCO₃⁻.
- Enhanced Third Dissociation: The low H⁺ concentration also influences the third dissociation step (Eq.3). Similar to the second step, the limited availability of H⁺ ions favours the dissociation of HCO₃⁻ to form carbonate ions (CO₃²⁻). This enhanced dissociation leads to a significant increase in the concentration of CO₃²⁻ in the solution.
Here's where CO₃²⁻ plays a starring role in elevating the pH. While CO₃²⁻ ions are classified as weak bases, they can interact with water molecules to a small extent, producing a limited amount of OH⁻ ions according to the following equation:
CO₃²⁻ + H₂O ⇌ HCO₃⁻ + OH⁻ (Eq.4)
However, it's crucial to understand that the formation of CO₃²⁻ itself is the primary driver of the high pH environment. These newly formed CO₃²⁻ ions have a remarkable ability to "mop up" any remaining H⁺ ions in the solution. By consuming H⁺ ions, CO₃²⁻ effectively reduces the concentration of H⁺, which is the very definition of what makes a solution acidic. This decrease in H⁺ concentration pushes the pH of the solution upwards, creating a more alkaline environment. Therefore, in essence, the low H⁺ concentration at high pH promotes the dissociation of carbonic acid, leading to a significant increase in CO₃²⁻ ions. These CO₃²⁻ ions, by consuming H⁺ ions, become the driving force behind the high pH (alkaline) nature of the solution.
The Iron Carbonate Layer: Structure and Protective Nature
The direct product of CO₂ corrosion is the formation of iron carbonate (FeCO₃). Fig.3 represents the steps of FeCO₃ formation.
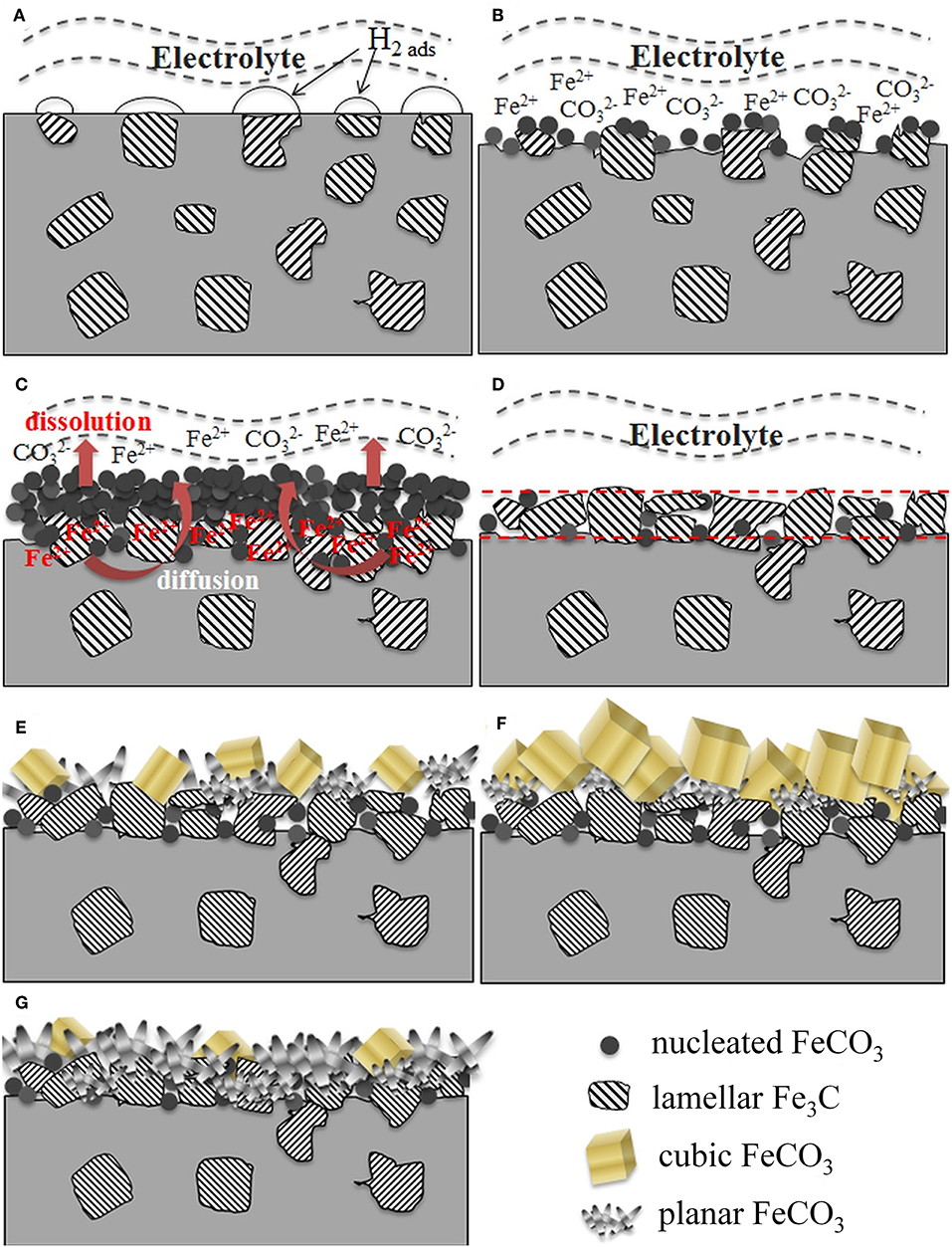 Fig.3:
Schematic diagrams showing the formation of corrosion scales on carbon steel immersed in CO2-saturated NaCl solutions as a function of time, temperature and salt concentration. (A) initial corrosion upon immersion; (B) early FeCO3 nucleation; (C) accumulation of FeCO3; (D) accumulation of Fe3C; (E) dual-layer formation; (F) growth of cubic FeCO3 in outer layer; (G) compact inner layer with planar FeCO3. (source: Li et al. (2019) front. Mater. 6:10. https://doi.org/10.3389/fmats.2019.00010 )
Fig.3:
Schematic diagrams showing the formation of corrosion scales on carbon steel immersed in CO2-saturated NaCl solutions as a function of time, temperature and salt concentration. (A) initial corrosion upon immersion; (B) early FeCO3 nucleation; (C) accumulation of FeCO3; (D) accumulation of Fe3C; (E) dual-layer formation; (F) growth of cubic FeCO3 in outer layer; (G) compact inner layer with planar FeCO3. (source: Li et al. (2019) front. Mater. 6:10. https://doi.org/10.3389/fmats.2019.00010 )
FeCO₃ adopts a rhombohedral crystal structure. This means the iron (Fe²⁺) cations occupy the centre of a distorted trigonal prism formed by six carbonate (CO₃²⁻) anions. Each carbonate anion, in turn, bonds to three iron cations through its three oxygen atoms. This arrangement creates a strong, three-dimensional network that contributes to the stability of the FeCO₃ layer. FeCO₃ usually formed as a protective layer, that can prevent further corrosion of steel surface.
Reasons for FeCO₃'s Protection:
- Barrier Effect: The FeCO₃ layer physically hinders the access of corrosive agents like carbonic acid (H₂CO₃) and dissolved oxygen (O₂) to the underlying iron surface. This reduces the rate of the initial corrosion reaction :
Fe (s) → Fe²⁺ (aq) + 2e⁻ [Eq.5]
- Passivation: FeCO₃ formation passivates the iron surface. Passivation refers to the formation of a protective layer that inhibits further corrosion. The tightly packed FeCO₃ layer creates a diffusion barrier, slowing down the movement of both iron ions outwards and aggressive ions inwards.
However, the FeCO₃ layer can be affected by some process factors such as pH and temperature, flow, turbulence, and other parameters. Some of these parameters will be discussed in the following sections.
The effect of pH :
- Low pH (Acidic): In acidic environments (low pH), the solubility of FeCO₃ increases. This means the FeCO₃ layer can dissolve more readily according to the following equilibrium reaction:
FeCO₃ (s) + H⁺ (aq) ⇌ Fe²⁺ (aq) + HCO₃⁻ (aq) [Eq.6]
As the H⁺ concentration increases (lower pH), the equilibrium shifts towards the right, favouring the dissolution of FeCO₃ and weakening the protective layer.
- High pH (Alkaline): While high pH (alkaline) environments generally reduce the solubility of FeCO₃, other factors come into play. At very high pH, the formation of stable iron hydroxide complexes (e.g., Fe(OH)₃) becomes more favourable. These hydroxide complexes can disrupt the integrity of the FeCO₃ layer, compromising its effectiveness.
The effect of temperature:
- Increased Temperature (Up to a Point): Generally, elevated temperatures (within a moderate range) can promote the formation of a denser and more adherent FeCO₃ layer. This is because higher temperatures increase the reaction rate between Fe²⁺ ions and CO₃²⁻ ions, leading to faster precipitation of FeCO₃ on the steel surface.
- Excessive Temperature: However, excessively high temperatures can have detrimental effects. At very high temperatures, the thermal decomposition of FeCO₃ becomes significant. This decomposition can be represented as:
FeCO₃ (s) → FeO (s) + CO₂ (g) [Eq.7]
The breakdown of FeCO₃ can lead to the formation of a less protective iron oxide (FeO) layer and the release of CO₂ gas, potentially causing cracks and gaps in the protective film.
The effect of Flow and Turbulence: The Double-Edged Sword for Iron Carbonate Layers
The stability of the iron carbonate (FeCO₃) layer is not just a story of pH and temperature. Flow velocity and turbulence in the CO₂-containing solution play a crucial role, introducing both chemical and physical effects that can weaken or even disrupt the protective layer. Let's delve deeper into this.
Mass Transfer and Depletion:
- Mass Transfer Equation: The rate of corrosion is indeed governed by the mass transfer principles. This equation describes the movement of chemical species from the bulk solution to the steel surface and vice versa. For FeCO₃ stability, the key species are:
Reactants: CO₂ (dissolved carbon dioxide) and H⁺ (protons) act as corrosive agents that need to reach the steel surface to initiate the corrosion reaction (Eq.5)
Product: Fe²⁺ (ferrous ions) are the product of the initial corrosion reaction and need to be removed from the surface for the reaction to continue.
- Flow Velocity and Depletion: Higher flow velocities enhance mass transfer in both directions. This consequently means:
Increased Delivery of Corrosive Agents: Faster flow brings more CO₂ and H⁺ ions to the steel surface, potentially overwhelming the FeCO₃ layer's ability to prevent their access.
Faster Removal of Fe²⁺: While beneficial for promoting the forward corrosion reaction, the rapid removal of Fe²⁺ ions can also have a negative effect. Fe²⁺ ions are essential for the continuous formation of FeCO₃ on the steel surface . With faster removal due to high flow, there might not be enough Fe²⁺ available for ongoing FeCO₃ production and layer repair, making it more susceptible to depletion.
Fe²⁺ (aq) + CO₃²⁻ (aq) → FeCO₃ (s) [Eq.8]
Turbulence: The Disruptive Force:
Turbulent flow adds another layer of complexity. Imagine eddies and swirling currents constantly buffeting the FeCO₃ layer. This turbulence can have several detrimental effects:
- Physical Disruption: The turbulent forces can directly erode the FeCO₃ layer, breaking off particles and exposing fresh steel surfaces to the corrosive environment. This weakens the overall integrity of the protective barrier.
- Enhanced Mass Transfer: Similar to high flow velocity, turbulence further intensifies the mass transfer processes. This leads to a faster depletion of Fe²⁺ ions from the steel surface, as explained previously.
- Mixing and Inhomogeneity: Turbulent flow can lead to an uneven distribution of reactants and products around the FeCO₃ layer. This can create localised "hot spots" where the concentration of corrosive agents is higher, potentially leading to accelerated corrosion in specific areas.
These combined effects of flow and turbulence make FeCO₃ vulnerable for two main reasons:
- Depletion and Incompleteness: High flow and turbulence can deplete the Fe²⁺ ions needed for ongoing FeCO₃ formation, potentially leaving the steel surface partially or fully exposed. This "patchy" protection weakens the overall effectiveness of the FeCO₃ layer.
- Physical Damage: Turbulent forces can directly damage the FeCO₃ layer, creating cracks and gaps that allow corrosive agents to reach the steel surface more easily, further accelerating corrosion.
Normally, CO2corrosion causes localized corrosion (Fig.4), which can take different forms, specially when the protective FeCO₃ layer is disrupted. Some of the common localized damages include:
- Pitting corrosion
- Mesa Attack
- Flow induced corrosion
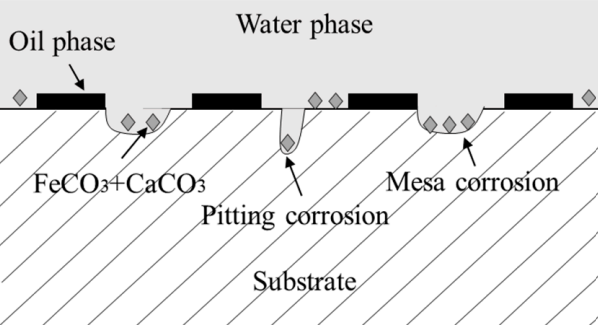 Fig.4: some forms of CO2 corrosion attacks. (source: Bai et al. 2019,
Materials 2018, 11, 1765; doi:10.3390/ma11091765)
Fig.4: some forms of CO2 corrosion attacks. (source: Bai et al. 2019,
Materials 2018, 11, 1765; doi:10.3390/ma11091765)
The Detrimental Role of Dissolved Oxygen in CO₂ Corrosion and Iron Carbonate Stability.
While the presence of CO₂ plays a key role in CO₂ corrosion, the influence of dissolved oxygen (O₂) cannot be ignored. The presence of O₂ significantly impacts the formation and stability of the protective iron carbonate (FeCO₃) layer, ultimately accelerating the corrosion rate in CO₂ environments. Let's shed some lights on the chemical and physical reasons behind this detrimental effect in the next sections.
Competitive Oxidation and Instability:
- Iron Oxidation: In the absence of O₂, the primary corrosion reaction involves the direct dissolution of iron (Fe) into ferrous ions (Fe²⁺), (Eq.5).
- Dissolved Oxygen's Interplay: When O₂ is present, it readily (much faster) reacts with ferrous ions to form ferric ions (Fe³⁺):
4Fe²⁺ (aq) + O₂ (aq) + 4H⁺ (aq) → 4Fe³⁺ (aq) + 2H₂O (l) [Eq.9]
- Impact on FeCO₃ Formation: Fe³⁺ ions do not readily participate in the formation of FeCO₃. This disrupts the crucial balance needed for a stable FeCO₃ layer. Ideally, Fe²⁺ ions from the initial corrosion reaction (Eq.5) should react with dissolved carbonate ions (CO₃²⁻) to form the protective layer (Eq.8)
However, with the presence of O₂, the conversion of Fe²⁺ to Fe³⁺ (Eq.9) reduces the availability of Fe²⁺ for FeCO₃ formation (Eq. 8). The conversion (oxidation of Fe²⁺) is significantly faster than its reaction with CO₃²⁻ to form FeCO₃, hindering the formation of a protective iron carbonate layer (Eqs 8&9).
Localized Corrosion and Destabilization:
- Localized Attack: The presence of O₂ can also lead to localized areas of intense corrosion. This occurs because O₂ can participate in a cathodic reaction, consuming electrons at specific sites on the steel surface. This localized consumption of electrons creates an uneven distribution of anodic (oxidation) and cathodic (reduction) reactions, leading to a more aggressive form of corrosion called pitting.
- Destabilization of FeCO₃: Even if some FeCO₃ forms, O₂ can react with it, leading to its destabilization. This reaction is represented by Eq.10:
4FeCO₃ (s) + O₂ (aq) + 6H₂O (l) → 4Fe(OH)₃ (s) + 8HCO₃⁻ (aq) [Eq. 10]
Here, FeCO₃ is converted to the less protective iron hydroxide (Fe(OH)₃). This conversion not only weakens the overall chemical composition of the protective layer but also introduces physical vulnerabilities.
The presence of iron oxides formed from this reaction (or from other pathways) can disrupt the physical integrity of the FeCO₃ layer. Iron oxides often have different crystal structures and packing compared to FeCO₃. When these oxides mix in with the FeCO₃ layer, they can create gaps and imperfections. This disrupts the barrier's ability to effectively hinder the access of corrosive agents to the underlying steel surface.
The Power of Stainless steel: Why Chromium Oxide Reigns Supreme
The superior resistance of 300 series stainless steel to CO₂ corrosion compared to carbon steel hinges on the presence of chromium (Cr) in its alloy. Let's delve deeper into why the chromium oxide (Cr₂O₃) layer it forms is significantly more robust than the iron carbonate (FeCO₃) layer on carbon steel, exploring both chemical and atomic bonding aspects:
Chemical Advantages of Chromium Oxide:
- Oxidation State: Chromium in Cr₂O₃ exists in the +3 oxidation state (Cr³⁺). This stable +3 state makes Cr₂O₃ less susceptible to reduction by common corrosive agents like H⁺ ions. In contrast, Fe²⁺ in FeCO₃ can be further oxidised to Fe³⁺ under certain conditions, potentially leading to the breakdown of the FeCO₃ layer.
- Ionic vs. Covalent Character: Cr₂O₃ exhibits a predominantly ionic bond between Cr³⁺ and O²⁻ ions. This ionic character contributes to the strong electrostatic attraction between the ions, leading to a more robust and tightly packed oxide layer. FeCO₃, on the other hand, has a more covalent character in its bonding between Fe²⁺ and CO₃²⁻. Covalent bonds, while strong, can be more susceptible to disruption under harsh conditions compared to ionic bonds.
Atomic Bonding and Crystal Structure:
- Cr₂O₃ Crystal Structure: Chromium oxide adopts a corundum crystal structure. In this structure, each Cr³⁺ ion is surrounded by six O²⁻ ions in a distorted octahedral arrangement. This highly symmetrical and densely packed arrangement promotes strong interatomic forces, leading to a stable and robust oxide layer.
- FeCO₃ Crystal Structure: Iron carbonate has a rhombohedral crystal structure. While it's a stable structure, the bonding between Fe²⁺ and CO₃²⁻ ions is not as strong or as directionally dependent as the ionic bonding in Cr₂O₃. This can lead to a more loosely packed and potentially more vulnerable structure compared to Cr₂O₃.
Synergistic Effect:
The combined effect of chromium's stable +3 oxidation state, the predominantly ionic character of Cr₂O₃ bonding, and its robust corundum crystal structure all contribute to a highly effective barrier against CO₂ corrosion. The Cr₂O₃ layer is much more resistant to reduction, diffusion of corrosive agents, and physical disruption compared to the FeCO₃ layer on carbon steel. This is why 300 series stainless steel offers superior protection in CO₂-containing environments.
Conclusion
CO₂ corrosion poses a significant threat to metallic infrastructure across various industries. Understanding the underlying chemical and physical processes, from CO₂ dissolution to the interplay between temperature, flow, and the protective FeCO₃ layer, is crucial for developing effective mitigation strategies. Material selection, corrosion inhibitors, and proper design considerations that minimize flow.

About the Author:
Dr. Mustafa Hashim
(PhD in Corrosion Engineering, NACE Senior Corrosion Technologist).
Dr Hashim is a corrosion expert with over 19 years of experience in the oil and gas industry. He has extensive experience in managing and controlling corrosion across various sectors, including refineries, petrochemical units, and power plants, tackling challenges in both upstream and downstream operations. His expertise encompasses mitigating corrosion threats in diverse environments, including those involving CO2
From Invisible Gas to Destructive Force: A Scientific Look at CO₂ Corrosion